Van der Waals force
In physical chemistry, the van der Waals force (or van der Waals interaction), named after Dutch scientist Johannes Diderik van der Waals, is the sum of the attractive or repulsive forces between molecules (or between parts of the same molecule) other than those due to covalent bonds or to the electrostatic interaction of ions with one another or with neutral molecules.[1] The term includes:
- force between two permanent dipoles (Keesom force)
- force between a permanent dipole and a corresponding induced dipole (Debye force)
- force between two instantaneously induced dipoles (London dispersion force)
It is also sometimes used loosely as a synonym for the totality of intermolecular forces. Van der Waals forces are relatively weak compared to normal chemical bonds, but play a fundamental role in fields as diverse as supramolecular chemistry, structural biology, polymer science, nanotechnology, surface science, and condensed matter physics. Van der Waals forces define the chemical character of many organic compounds. They also define the solubility of organic substances in polar and non-polar media. In low molecular weight alcohols, the properties of the polar hydroxyl group dominate the weak intermolecular forces of van der Waals. In higher molecular weight alcohols, the properties of the nonpolar hydrocarbon chain(s) dominate and define the solubility. Van der Waals-London forces grow with the length of the nonpolar part of the substance.
Contents |
Definition
Van der Waals forces include attractions between atoms, molecules, and surfaces, as well as other intermolecular forces. They differ from covalent and ionic bonding in that they are caused by correlations in the fluctuating polarizations of nearby particles (a consequence of quantum dynamics[2]).
Intermolecular forces have four major contributions:
- A repulsive component resulting from the Pauli exclusion principle that prevents the collapse of molecules.
- Attractive or repulsive electrostatic interactions between permanent charges (in the case of molecular ions), dipoles (in the case of molecules without inversion center), quadrupoles (all molecules with symmetry lower than cubic), and in general between permanent multipoles. The electrostatic interaction is sometimes called the Keesom interaction or Keesom force after Willem Hendrik Keesom.
- Induction (also known as polarization), which is the attractive interaction between a permanent multipole on one molecule with an induced multipole on another. This interaction is sometimes called Debye force after Peter J.W. Debye.
- Dispersion (usually named after Fritz London), which is the attractive interaction between any pair of molecules, including non-polar atoms, arising from the interactions of instantaneous multipoles.
Returning to nomenclature, different texts refer to different things using the term "van der Waals force". Some texts mean by the van der Waals force the totality of forces (including repulsion); others mean all the attractive forces (and then sometimes distinguish van der Waals-Keesom, van der Waals-Debye, and van der Waals-London).
All intermolecular/van der Waals forces are anisotropic (except those between two noble gas atoms), which means that they depend on the relative orientation of the molecules. The induction and dispersion interactions are always attractive, irrespective of orientation, but the electrostatic interaction changes sign upon rotation of the molecules. That is, the electrostatic force can be attractive or repulsive, depending on the mutual orientation of the molecules. When molecules are in thermal motion, as they are in the gas and liquid phase, the electrostatic force is averaged out to a large extent, because the molecules thermally rotate and thus probe both repulsive and attractive parts of the electrostatic force. Sometimes this effect is expressed by the statement that "random thermal motion around room temperature can usually overcome or disrupt them" (which refers to the electrostatic component of the van der Waals force). Clearly, the thermal averaging effect is much less pronounced for the attractive induction and dispersion forces.
The Lennard-Jones potential is often used as an approximate model for the isotropic part of a total (repulsion plus attraction) van der Waals force as a function of distance.
Van der Waals forces are responsible for certain cases of pressure broadening (van der Waals broadening) of spectral lines and the formation of van der Waals molecules. The London-van der Waals forces are related to the Casimir effect for dielectric media, the former being the microscopic description of the latter bulk property. The first detailed calculations of this were done in 1955 by E. M. Lifshitz.[3][4]
London dispersion force
London dispersion forces, named after the German-American physicist Fritz London, are weak intermolecular forces that arise from the interactive forces between instantaneous multipoles in molecules without permanent multipole moments. These forces dominate the interaction of non-polar molecules, and also play a less significant role in Van der Waals forces than molecules containing permanent dipoles or ionized molecules. London dispersion forces are also known as dispersion forces, London forces, or instantaneous dipole–induced dipole forces. They increase with the molar mass, causing a higher boiling point especially for the halogen group.
Van der Waals forces between macroscopic objects
For macroscopic bodies with known volumes and numbers of atoms or molecules per unit volume, the total van der Waals force is often computed based on the "microscopic theory" as the sum over all interacting pairs. It is necessary to integrate over the total volume of the object, which makes the calculation dependent on the objects' shapes. For example, the van der Waals interaction energy between spherical bodies of radii R1 and R2 and with smooth surfaces was approximated in 1937 by Hamaker[5] (using London's famous 1937 equation for the dispersion interaction energy between atoms/molecules[6] as the starting point) by:
-
![\ U(z;R_{1},R_{2})= -\frac{A}{6}\left[\frac{2R_{1}R_{2}}{z^2-(R_{1}%2B R_{2})^2} %2B \frac{2R_{1}R_{2}}{z^2-(R_{1}-R_{2})^2} %2B \ln\left(\frac{z^2-(R_{1}%2B R_{2})^2}{z^2-(R_{1}- R_{2})^2}\right)\right]](/2012-wikipedia_en_all_nopic_01_2012/I/d38927e3c3ccc70e1c758eec745c21ff.png)
(
where A is the Hamaker coefficient, which is a constant (~10−19 - 10−20 J) that depends on the material properties (it can be positive or negative in sign depending on the intervening medium), and z is the center-to-center distance, i.e. the sum of R1, R2, and r (the distance between the surfaces): 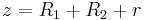 .
.
In the limit of close-approach, the spheres are sufficiently large compared to the distance between them, i.e. 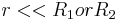 , so that equation (1) for the potential energy function simplifies to:
, so that equation (1) for the potential energy function simplifies to:
-
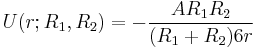
(
The van der Waals force between two spheres of constant radii (R1 and R2 are treated as parameters) is then a function of separation since the force on an object is the negative of the derivative of the potential energy function,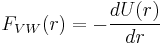 . This yields:
. This yields:
-
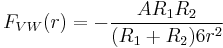
(
The van der Waals forces between objects with other geometries using the Hamaker model have been published in the literature.[7][8][9]
From the expression above, it is seen that the van der Waals force decreases with decreasing particle size (R). Nevertheless, the strength of inertial forces, such as gravity and drag/lift, decrease to a greater extent. Consequently, the van der Waals forces become dominant for collections of very small particles such as very fine-grained dry powders (where there are no capillary forces present) even though the force of attraction is smaller in magnitude than it is for larger particles of the same substance. Such powders are said to be cohesive, meaning they are not as easily fluidized or pneumatically conveyed as easily as their more coarse-grained counterparts. Generally, free-flow occurs with particles greater than about 250 μm.
The van der Waals force of adhesion is also dependent on the surface topography. If there are surface asperities, or protuberances, that result in a greater total area of contact between two particles or between a particle and a wall, this increases the van der Waals force of attraction as well as the tendency for mechanical interlocking.
The microscopic theory assumes pairwise additivity. It neglects many-body interactions and retardation. A more rigorous approach accounting for these effects, called the "macroscopic theory," was developed by Lifshitz in 1956.[10] Langbein derived a much more cumbersome "exact" expression in 1970 for spherical bodies within the framework of the Lifshitz theory[11] while a simpler macroscopic model approximation had been made by Derjaguin as early as 1934.[12] Expressions for the van der Waals forces for many different geometries using the Lifshitz theory have likewise been published.
Use by animals
The ability of geckos – which can hang on a glass surface using only one toe – to climb on sheer surfaces has been attributed to the van der Waals forces between these surfaces and the spatula (plural spatulae), or microscopic projections, which cover the hair-like setae found on their footpads.[13][14] A later study suggested that capillary adhesion might play a role,[15] but that hypothesis has been rejected by more recent studies.[16] [17] [18] There were efforts in 2008 to create a dry glue that exploits the effect,[19] and success was achieved in 2011 to create an adhesive tape on similar grounds.[20] In 2011, a paper was published relating the effect to both velcro-like hairs and the presence of lipids in gecko footprints.[21]
References
- ^ IUPAC, Compendium of Chemical Terminology, 2nd ed. (the "Gold Book") (1997). Online corrected version: (1994) "Van der Waals forces".
- ^ A.A. Abrikosov, L.P. Gorkov, I.E. Dzyaloshinsky (1963–1975). Methods of Quantum Field Theory in Statistical Physics. Dover Publications. ISBN 0-486-63228-8. Chapter 6 Electromagnetic Radiation in an Absorbing Medium
- ^ Dzyaloshinskii, I E; Lifshitz, E M; Pitaevskii, Lev P (1961). "GENERAL THEORY OF VAN DER WAALS' FORCES". Soviet Physics Uspekhi 4 (2): 153. Bibcode 1961SvPhU...4..153D. doi:10.1070/PU1961v004n02ABEH003330.
- ^ For further investigation, one may consult the University of St. Andrews' levitation work in a popular article: Science Journal: New way to levitate objects discovered, and in a more scholarly version: New Journal of Physics: Quantum levitation by left-handed metamaterials, which relate the Casimir effect to the gecko and how the reversal of the Casimir effect can result in physical levitation of tiny objects.
- ^ H. C. Hamaker, Physica, 4(10), 1058-1072 (1937)
- ^ F. London, Transactions of the Faraday Society, 33, 8-26 (1937)
- ^ R. Tadmor, JOURNAL OF PHYSICS: CONDENSED MATTER, 13 (2001) L195–L202
- ^ Israelachvili J., Intermolecular and Surface Forces, Academic Press (1985–2004), ISBN 0-12-375181-0
- ^ V. A. Parsegian, "Van der Waals Forces: A Handbook for Biologists, Chemists, Engineers, and Physicists," Cambridge University Press (2006) ISBN 978-0-521-83906-8
- ^ E. M. Lifshitz, Soviet Phys. JETP, 2, 73 (1956)
- ^ D. Langbein, Phys. Rev. B, 2, 3371 (1970)
- ^ B. V. Derjaguin, Kolloid-Z., 69, 155-64 (1934)
- ^ Researchers discover how geckos know when to hold tight. Clemson.edu. Retrieved on 2011-01-08.
- ^ Autumn, K. et al. (2002). "Evidence for van der Waals adhesion in gecko setae". Proceedings of the National Academy of Sciences 99 (19): 12252–6. doi:10.1073/pnas.192252799. PMC 129431. PMID 12198184. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=129431.
- ^ Huber, G., et al. (2005). "Evidence for capillarity contributions to gecko adhesion from single spatula nanomechanical measurements". Proceedings of the National Academy of Sciences 102 (45): 16293–6. doi:10.1073/pnas.0506328102. PMC 1283435. PMID 16260737. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1283435.
- ^ Chen, B.; Gao, H. (2010). "An alternative explanation of the effect of humidity in gecko adhesion: stiffness reduction enhances adhesion on a rough surface". Int JAppl Mech 2: 1–9. doi:10.1142/s1758825110000433.
- ^ Puthoff, J. B., et al. (2010). "Changes in materials properties explain the effects of humidity on gecko adhesion". J Exp Biol 213 (21): 3699–3704. doi:10.1242/jeb.047654.
- ^ Prowse, M. S., et al. (2011). "Effects of humidity on the mechanical properties of gecko setae". Acta Biomater 7 (2): 733–738. doi:10.1016/j.actbio.2010.09.036. PMID 20920615.
- ^ Gecko-like glue is said to be stickiest yet, "reuters.com" 8 October 2008
- ^ http://www.gizmag.com/bioinspired-adhesive-tape-kiel/20406/
- ^ Scientists trace gecko footprint, find clue to glue
..
Further reading
- Iver Brevik, V. N. Marachevsky, Kimball A. Milton, Identity of the Van der Waals Force and the Casimir Effect and the Irrelevance of these Phenomena to Sonoluminescence, hep-th/9901011
- I. D. Dzyaloshinskii, E. M. Lifshitz, and L. P. Pitaevskii, Usp. Fiz. Nauk 73, 381 (1961)
- English translation: Soviet Phys. Usp. 4, 153 (1961)
- L. D. Landau and E. M. Lifshitz, Electrodynamics of Continuous Media, Pergamon, Oxford, 1960, pp. 368–376.
- Dieter Langbein, "[Langbein, Dieter Theory of Van der Waals Attraction, ( Springer-Verlag New York Heidelberg 1974)]"
- Mark Lefers, "Van der Waals dispersion force". Holmgren Lab.
- E. M. Lifshitz, Zh. Eksp. Teor. Fiz. 29, 894 (1955)
- English translation: Soviet Phys. JETP 2, 73 (1956)
- Western Oregon University's "London force". Intermolecular Forces. (animation)
- J. Lyklema, Fundamentals of Interface and Colloid Science, page 4.43
- Israelachvili J., Intermolecular and Surface Forces, Academic Press (1985–2004), ISBN 0-12-375181-0
External links
- Senese, Fred (1999). "What are van der Waals forces?". Frostburg State University. http://antoine.frostburg.edu/chem/senese/101/liquids/faq/h-bonding-vs-london-forces.shtml. Retrieved March 2010. An introductory description of the van der Waals force (as a sum of attractive components only)
- Robert Full: Learning from the gecko's tail. TED Talk on biomimicry, including applications of Van der Waals force.
|
||||||||||||||||||||||||